

五步获得医疗器械FDA认证,FDA或将大幅度简化医械上市路径
时间:2018-12-11 11:39:56 作者:临测检测技术(上海)有限公司 点击:
次
美国的医疗器械市场是全球大的市场,2016年的数据显示美国的医疗器械市场规模占全球的51%,达到2084亿美元,对于医疗器械的企业来说是非常有吸引力的。医疗器械要进入美国市场,首先就要获得美国食品药品监督局(FDA)的认证。今天,小编与大家分享的是医疗器械要通过FDA认证,在上市前要经历的五个步骤。CE认证医疗器械通过FDA认证主要有5个步骤
确定器械的分类
选择正确的上市前申请途径
准备适当的上市前申请的信息
递交信息给FDA并与之沟通
企业注册和产品列名
如果你的产品是要满足QSR 820的要求的,那在上市前你的质量管理体系也要按照QSR 820的要求准备好, 对于大部分的II类器械(要求是510(k)), FDA在上市前不会到制造商现场进行审核。对于三类器械(要求是PMA), FDA会在上市前到制造商现场进行审核。
步骤一:确定器械分类
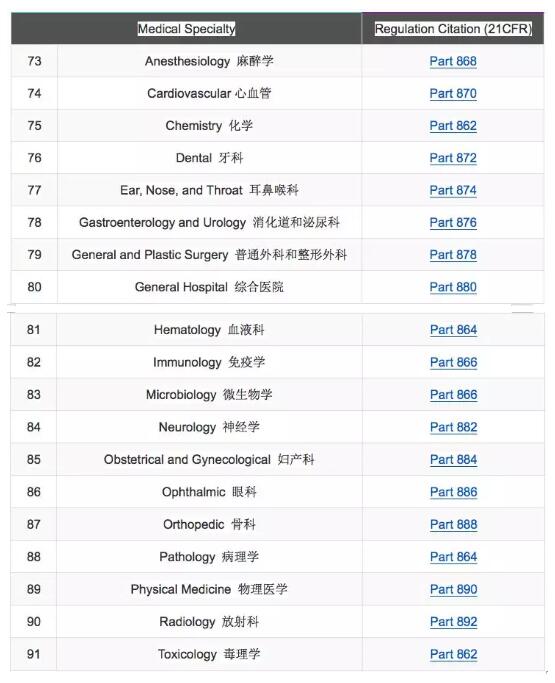
同我国的器械分类的基本原则一样,FDA的分类原则也是基于器械的风险。根据风险的大小不同,FDA将器械分为三类,Class I,Class II和Class III,并将1700多种不同类型的器械划分到16个医疗专业组中。确定器械的分类时,可以按照16个医疗专业组去找器械对应的分类,请见下表。
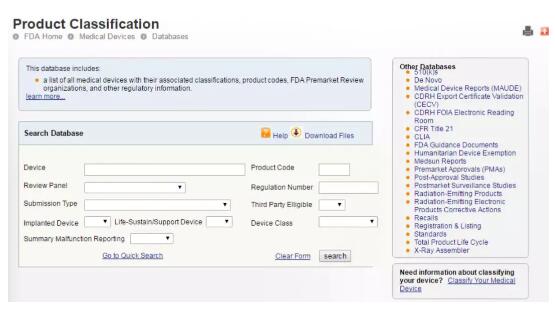
另外,还可以利用医疗器械分类数据库,直接搜索器械的名称查找分类,请见下图。
步骤二:选择正确的上市前申请途径
确定了产品分类后,根据法规的要求选择上市前申请的途径,主要包括四种:
510(k) (Premarket Notification):小部分Class I和大部分Class II需要采取的途径
PMA (Premarket Approval):大部分Class III需要采取的途径
De Novo (Evaluation of Automatic Class III Designation):新类型且风险程度不高的医疗器械可以采取的途径。
HDE (Humanitarian Device Exemption):罕见病所用到的器械采取的途径[D1] ,比如:治疗多发性骨髓瘤患者用的人造肾脏。
步骤三:准备适当的上市前申请的信息
上市前申请的信息总的来说包括以下四个方面:
设计控制:所有Class II和Class III器械必须按照质量体系法规(21 CFR 820.30)的设计控制进行设计。
非临床测试:基于器械的操作性能、材料等在GLP实验室检测。
临床证据:PMA, HDE、一些510(k)和De Novo需要临床证据。
标识:FFDCA Section 201(k) 定义的标识包括标签、宣传册、用户手册、说明书等
步骤四:递交信息给FDA并与之沟通
准备好递交的信息,便可将材料递交给FDA。我们以510(k)为例,在提交时需要同时提交两份资料,一份为电子版eCopy,一份是纸质版。提交资料审查之前,需先完成缴费。医疗器械的申请途径不同,FDA的收费也不同。 请看下图:
FDA在收到资料并确认企业已完成缴费,便会对资料后进行审查。审查过程中,如果FDA对企业提交的资料有疑问或者需要发补,审查的计时会暂停。在这个期间,与FDA的官员保持良好的沟通是非常必要的。以510(k)为例,审查期间的沟通时间表请见下图:
步骤五:企业注册和产品列名
审核通过后,企业距离将产品销售到美国市场就差一步了,这就企业注册和产品列名。21 CFR Part 807规定,所有在美国使用的医疗设备的生产和分销涉及的机构的所有者或经营者必须每年向FDA注册。因此企业需要向FDA缴纳年度注册费。下表是2018财年与2019财年的注册费用。
Year
FY 2018
FY 2019
Fee
$4,624
$4,884
产品列名是需要企业在网上提交产品的相关信息,告诉FDA即将在美国市场销售该产品。
医疗器械上市路径要简化?美FDA将大幅修改510(k)法规
美国FDA目前使用的510(k)法规最初是在1976年的医疗器械修正案中确立的,并在1990年的《安全医疗器械法》中进行了修订。510(k)是向FDA递交的上市前申请文件,目的是证明申请上市的器械与不受上市前批准影响(PMA)的合法上市器械同样安全有效。申请者必须把申请上市的器械与现在美国市场一种或多种相似器械对比,得出支持同样安全有效的结论。总体上,外界对510(k)审批程序一直非常关注。不过,日前有迹象显示,FDA或将大幅度修改这一法案。
2018年11月26日,美国FDA局长Scott Gottlieb博士和设备与放射健康中心主任Jeffrey Shuren博士宣布,FDA正计划对510(k)流程按现阶段的实际情况进行修正。二人在关于FDA 510(k)计划现代化改革新步骤的声明中提到,此次推进对医疗器械安全性和有效性的审查声明,主要侧重于确保通过510(k)流程的新设备能够符合不断发展的安全性和有效性标准。
具体的关键点总结如下:
1、无需与老旧的、已获批准类似器械比较
声明指出,510(k)申请获批的产品中,有20%是与已获批准10年以上的老旧产品进行比较。这显然并不能代表当前快速发展和改进的医疗设备以及当前的临床水平。FDA认为,申请时应与更现代化的新设备进行利益和风险的比较。为此,FDA将推动使用更新的类似器械比较。FDA将在其网站上公布那些已证明是通过与老旧设备实质等同而最终获批的设备清单。
但是,目前还不清楚这个列表是否只包括最近获准的产品,或者注释整个510(k)数据库。此外,也不清楚如何处理多个设备实质等同的情况(比如与三个设备进行实质等同比较,但其中只有一个超过10年)。根据FDA的说法,“在上市前申请时推进使用新的设备进行比较,将让患者和医生在一种设备的旧版本和新版本中做出选择,促进更大的竞争,达到提高安全性和性能的功能,并帮助确保更新设备反映了更多现代技术和标准,可以改善患者护理和结果。”
2、淘汰落后的老旧类似器械
为了进一步消除使用老旧类似器械的目标,FDA表示,它将开始淘汰部分老旧的510(k)批准。目前尚不清楚这意味着什么,具体将如何运作,或者FDA是否具有相关的法定权力。举例来说,FDA指出,其正在努力消除某些引起安全问题的获批设备再次用于类比使用。尽管,此举清楚地表明针对的是已获批准的510(k)设备,但该过程也可能影响此前免于510(k)流程的其他设备。
新法规的修改或将改变特定设备类型510(k)豁免的界限。如果FDA对此进行彻底改变,此前510(k)豁免的条件和限制或将发生改变,这也是该项提案中必须解决的众多细节问题之一。
3、改进510(k)安全和性能标准评估途径
根据该声明,FDA还计划创建一个替代510(k)的途径,允许赞助商通过证明设备符合相关设备类型的安全和性能标准,来论证其具有实质等同性。FDA表示,这条新途径将基于当代水平评估新设备的水准,而不是与过时的设备进行比较。FDA在声明中表示,“我们相信这种方法是510(k)计划的未来,而不是一味地将过去获批的设备视为安全性和有效性的基准,甚至依赖超过数十年的老旧设备作为批准的参照物。FDA上市前审查将基于展望未来的现代水准和基线并可以随着技术的进步而保持更新。有时候,与老旧获批的类似设备进行比较,实际上会导致更先进的技术获得批准,导致病患丧失了新的治疗选择,因为创新型的技术产品很难与过时数十年的设备在比较中完成高度的结合或相似。”EMC测试
虽然声明中将此方法称为新的途径,但听起来很像当前的510(k)路径的简化版,它允许提交者主要依赖于符合共识标准的基线水平进行类比论证。简化的510(k)路径适用于某些设备类型,但对于大多数设备类型继续使用标准510(k)申请,并保持次标准更新以确保设备技术保持现代化。FDA认为,此举可以更容易向付款人证明他们的产品比市场上的其他设备表现更好,来帮助支持医保覆盖决策。
文章与图片来源网络,仅供技术交流。如有侵权,请通知我们删除。


